Here I present a proposal for original experimental research, aimed at elucidating the protein-protein interactions of a putative glycolytic metabolon in yeast. Through this I highlight a fascinating type of metabolic organization, metabolons. I also emphasize the utility of the Förster resonance energy transfer (FRET) technique for quantifying protein-protein interactions. Lastly, I stress the importance of experimental design and the proper use of controls.
Abstract
Many metabolic pathways are arranged in metabolons, where consecutive enzymes physically interact and metabolites are channeled directly from one enzyme to the next. In Saccharomyces cerevisiae (yeast), there is in vitro evidence that glycolysis is organized as a metabolon. However, the protein-protein interactions comprising this putative metabolon have yet to be characterized. The proposed research aims to study these interactions in vitro and in vivo. This will be done using a FRET-based strategy where consecutive enzymes will be tagged with cyan fluorescent protein (CFP) and yellow fluorescent protein (YFP) as the FRET donor and acceptor, respectively. In addition, the interactions of filamentous F-actin with these enzymes will be studied using fluorescence recovery after photobleaching (FRAP) experiments. Lastly, F-actin’s contribution to the stability of the metabolon will be elucidated. This research will increase our understanding of how the glycolytic pathway functions and is organized in vivo. Furthermore, the FRET-based probes developed may be useful tools towards further in vivo study of the glycolytic metabolon and its dynamic association as a means of metabolic regulation.
Introduction
Metabolic pathways are increasingly being found to be organized in metabolons, where participating enzymes physically interact [1]. This arrangement is advantageous by allowing substrate channeling between sequential enzymes along the pathway. This protects unstable/toxic intermediates, overcomes diffusive kinetic barriers, and limits use of the metabolites by other pathways [2]. Lastly, regulation of metabolic flux through mediation of these dynamic protein-protein interactions is an intriguing possibility.
Glycolysis involves the conversion of glucose to pyruvate with energy generation in the form of ATP [3]. This metabolic pathway is achieved using ten enzymes (Fig. 1):
Glycolysis involves the conversion of glucose to pyruvate with energy generation in the form of ATP [3]. This metabolic pathway is achieved using ten enzymes (Fig. 1):
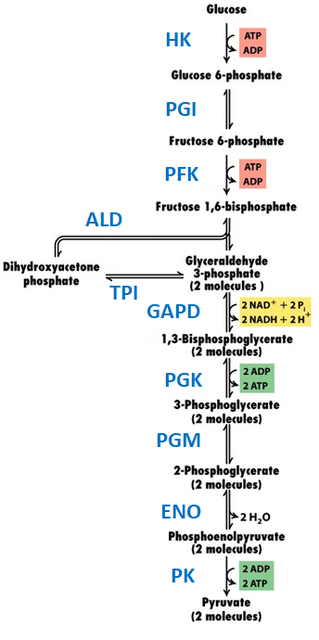
Figure 1. Glycolysis pathway. HK–hexokinase, PGI–phosphoglucoisomerase, PFK– phosphofructokinase, ALD–aldolase, TPI–triose phosphate isomerase, GAPD–glyceraldehyde phosphate dehydrogenase, PGK–phosphoglycerokinase, PGM–phosphoglyceromutase, ENO–enoloase, PK–pyruvate kinase. Figure modified from: Molecular cell biology (2008), New York: W.H. Freeman and Co.
Being the basis of both aerobic and anaerobic respiration, glycolysis is one of the most fundamental metabolic pathways. In fact, it is present in most known organisms [3]. Understanding the organization and regulation of glycolysis is therefore vital to understanding metabolism. There is now significant evidence that glycolytic enzymes associate as a metabolon. This has been well-studied in mammalian muscle cells, erythrocytes, and bacteria [3,4]. However, most of this evidence comes from in vitro work, including kinetic studies and characterization of protein-protein interactions. Recent reviews have highlighted a need for in vivo study and validation of this putative metabolon [3].
Of all model organisms, the glycolytic metabolon of S. cerevisiae is surprisingly under-studied. Recent work by Araiza-Olivera et al. provided the first strong evidence for a glycolytic metabolon in yeast [5]. The glycolytic activity of yeast cells permeabilized with streptolysin O was found to be resistant to inhibition by targeted antibodies or viscogens. In contrast, the isolated glycolytic enzymes were shown to be susceptible to inhibition by these agents. This kinetic protection is a typical feature of metabolons. Furthermore, it is proposed that this metabolon interacts directly with and is stabilized by filamentous F-actin. In support of this, ALD and GAPD were shown to bind yeast F-actin in co-sedimentation experiments [6]. More recently, co-immunoprecipitation experiments indicated that all but three (PFK, PGM and PK) of the ten yeast glycolytic enzymes bind F-actin [5]. In contrast, none of them co-immunoprecipitated with monomeric G-actin. Lastly, disruption of F-actin by cytochalasin D reduced the rate of glycolysis supported by a yeast cytoplasmic extract [5]. This data makes a strong case for the existence of a glycolytic metabolon in S. cerevisiae. However, several deficiencies in the current body of evidence are apparent. Firstly, evidence for the expected protein-protein interactions between sequential enzymes of the pathway is currently lacking. Secondly, despite compelling indirect evidence, it has not yet been explicitly shown that F-actin stabilizes the metabolon’s protein-protein interactions. Lastly, there is currently no in vivo evidence of a glycolytic metabolon in S. cerevisiae, all studies to date have used in vitro methods. To address these limitations, the proposed research aims to characterize the protein-protein interactions between the enzymes of the glycolytic metabolon of S. cerevisiae in vitro and in vivo. Furthermore, the contribution of F-actin to the stability of this complex will be elucidated. Based on the current literature evidence, a model for the S. cerevisiae glycolytic metabolon complex is proposed in Fig. 2.
Being the basis of both aerobic and anaerobic respiration, glycolysis is one of the most fundamental metabolic pathways. In fact, it is present in most known organisms [3]. Understanding the organization and regulation of glycolysis is therefore vital to understanding metabolism. There is now significant evidence that glycolytic enzymes associate as a metabolon. This has been well-studied in mammalian muscle cells, erythrocytes, and bacteria [3,4]. However, most of this evidence comes from in vitro work, including kinetic studies and characterization of protein-protein interactions. Recent reviews have highlighted a need for in vivo study and validation of this putative metabolon [3].
Of all model organisms, the glycolytic metabolon of S. cerevisiae is surprisingly under-studied. Recent work by Araiza-Olivera et al. provided the first strong evidence for a glycolytic metabolon in yeast [5]. The glycolytic activity of yeast cells permeabilized with streptolysin O was found to be resistant to inhibition by targeted antibodies or viscogens. In contrast, the isolated glycolytic enzymes were shown to be susceptible to inhibition by these agents. This kinetic protection is a typical feature of metabolons. Furthermore, it is proposed that this metabolon interacts directly with and is stabilized by filamentous F-actin. In support of this, ALD and GAPD were shown to bind yeast F-actin in co-sedimentation experiments [6]. More recently, co-immunoprecipitation experiments indicated that all but three (PFK, PGM and PK) of the ten yeast glycolytic enzymes bind F-actin [5]. In contrast, none of them co-immunoprecipitated with monomeric G-actin. Lastly, disruption of F-actin by cytochalasin D reduced the rate of glycolysis supported by a yeast cytoplasmic extract [5]. This data makes a strong case for the existence of a glycolytic metabolon in S. cerevisiae. However, several deficiencies in the current body of evidence are apparent. Firstly, evidence for the expected protein-protein interactions between sequential enzymes of the pathway is currently lacking. Secondly, despite compelling indirect evidence, it has not yet been explicitly shown that F-actin stabilizes the metabolon’s protein-protein interactions. Lastly, there is currently no in vivo evidence of a glycolytic metabolon in S. cerevisiae, all studies to date have used in vitro methods. To address these limitations, the proposed research aims to characterize the protein-protein interactions between the enzymes of the glycolytic metabolon of S. cerevisiae in vitro and in vivo. Furthermore, the contribution of F-actin to the stability of this complex will be elucidated. Based on the current literature evidence, a model for the S. cerevisiae glycolytic metabolon complex is proposed in Fig. 2.
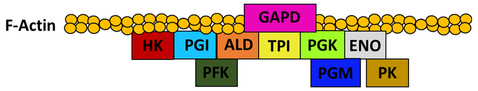
Figure 2. Proposed model of protein-protein interactions in the glycolytic metabolon of S. cerevisiae.
Proposal – Experiments
Interactions between glycolytic enzymes in vitro
Binary interactions between sequential glycolytic enzymes will be characterized in vitro using a FRET-based strategy (Fig. 3). Sequential glycolytic enzyme pairs will be tagged with a CFP (FRET donor) and YFP (FRET acceptor) respectively. If the two proteins interact in vitro, then titration of the acceptor-tagged protein into a solution of the donor will cause a shift in its fluorescence emission spectrum due to a FRET interaction. It will be possible to obtain a binding curve from these data and calculate dissociation constants for each interaction. To validate observed interactions using an orthogonal method, co-immunoprecipitation will be performed using commercially available antibodies. All antibodies will be independently validated.
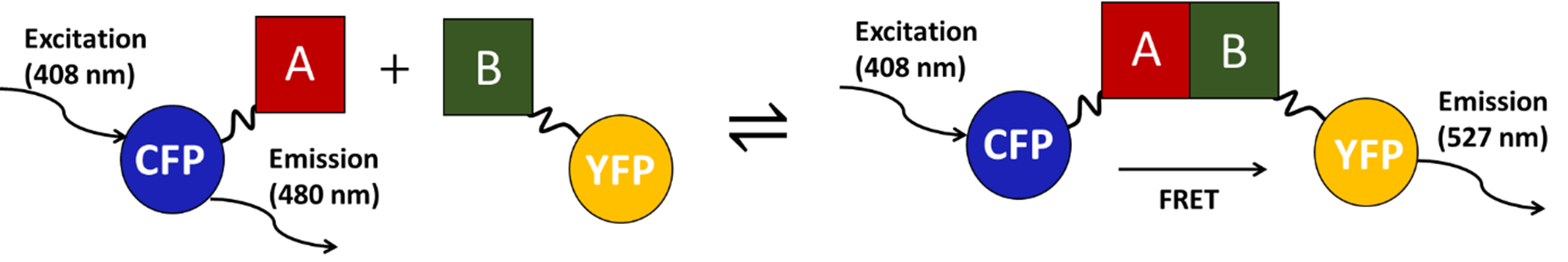
Figure 3. Quantitative detection of protein-protein interactions (between A and B) by FRET-based approach.
This FRET-based approach has several advantages over other techniques available to characterize protein-protein interactions. Unlike many techniques, it is suitable for characterization both in vitro and in vivo [7]. Furthermore, the FRET interaction is reversible and highly dependent on distance, making false positives unlikely. In contrast, bimolecular fluorescence complementation is more sensitive than FRET, but the interaction is irreversible [8]. This makes false positives a problem and limits the types of experiments that can be done. The yeast-2-hybrid assay is another approach to study protein-protein interactions in vivo [9]. However, this assay reports on interactions in the nucleus, while the interactions of interest occur in the cytoplasm and may require F-actin for stability. Furthermore, metabolon protein-protein interactions are generally of relatively low affinity [10]. Therefore, tandem affinity purification, which requires robust interactions, is also not a viable option.
Several control experiments are required to validate the FRET-based approach. As negative controls, the interaction of CFP and YFP alone will be tested in the above experiment. This will verify that interactions observed are not due to non-specific interactions between the fluorescent proteins. As an additional negative control, the interaction between glycolytic enzymes and Hog1 will be studied. Hog1 is known not to interact with yeast glycolytic enzyme [5]. We expect interactions between all enzymes which are consecutive in the metabolic pathway. A direct physical interaction is essential to allow substrate channeling. If any of these interactions are not detected by this assay, there are several alternative explanations. Firstly, F-actin may be essential to stabilize the interaction. If this is the case, it will be clarified when the experiment is repeated in the presence of F-actin, as described below. The interaction may also fail to be detected due to the design of the fusion proteins. The fluorescent protein tags may interfere with the proper folding of the enzymes, or may block the interaction face. This problem may be resolved by switching the position of the fluorescent protein tag on either protein (N or C-terminus), or by adjusting the linker length.
This FRET-based approach has several advantages over other techniques available to characterize protein-protein interactions. Unlike many techniques, it is suitable for characterization both in vitro and in vivo [7]. Furthermore, the FRET interaction is reversible and highly dependent on distance, making false positives unlikely. In contrast, bimolecular fluorescence complementation is more sensitive than FRET, but the interaction is irreversible [8]. This makes false positives a problem and limits the types of experiments that can be done. The yeast-2-hybrid assay is another approach to study protein-protein interactions in vivo [9]. However, this assay reports on interactions in the nucleus, while the interactions of interest occur in the cytoplasm and may require F-actin for stability. Furthermore, metabolon protein-protein interactions are generally of relatively low affinity [10]. Therefore, tandem affinity purification, which requires robust interactions, is also not a viable option.
Several control experiments are required to validate the FRET-based approach. As negative controls, the interaction of CFP and YFP alone will be tested in the above experiment. This will verify that interactions observed are not due to non-specific interactions between the fluorescent proteins. As an additional negative control, the interaction between glycolytic enzymes and Hog1 will be studied. Hog1 is known not to interact with yeast glycolytic enzyme [5]. We expect interactions between all enzymes which are consecutive in the metabolic pathway. A direct physical interaction is essential to allow substrate channeling. If any of these interactions are not detected by this assay, there are several alternative explanations. Firstly, F-actin may be essential to stabilize the interaction. If this is the case, it will be clarified when the experiment is repeated in the presence of F-actin, as described below. The interaction may also fail to be detected due to the design of the fusion proteins. The fluorescent protein tags may interfere with the proper folding of the enzymes, or may block the interaction face. This problem may be resolved by switching the position of the fluorescent protein tag on either protein (N or C-terminus), or by adjusting the linker length.
Protein production/purification
Expression vectors containing the open-reading frame for the S. cerevisiae glycolytic enzyme of interest followed by a linker, the fluorescent protein (YFP or CFP), and a His-tag will be prepared. The proteins will be recombinantly over-expressed in yeast. The His-tag will allow protein purification using metal affinity chromatography. Purity and identity will be verified by SDS-PAGE. Yeast is the ideal protein production system since this facilitates endogenous folding and proper post-translational modifications.
F-actin binding of glycolytic enzymes in vitro
Fluorescence recovery after photobleaching (FRAP) will be used to study binding between each glycolytic enzyme and F-actin filament in vitro. FRAP will be performed on each fluorescent protein-tagged enzyme in isolation and in the presence of excess F-actin. If the enzymes bind to F-actin, a large decrease in their diffusion rates is expected and therefore also a decrease in the fluorescence recovery rates. This will allow corroboration of the co-immunoprecipitation results of Araiza-Olivera et al. [5]. Further, it will verify that the fluorescent protein tags do not disrupt F-actin binding. Yeast actin is not commercially available but can be purified in good yield from yeast culture using the method of Waingeh et al. [6].
To ensure that changes in the FRAP recovery rates are the consequence of specific interactions between the glycolytic enzymes and F-actin, CFP and YFP will be used as negative controls. To further verify the specificity, the previously mentioned Hog1-fluoresent protein tagged construct can also be used as a negative control. All glycolytic enzymes are expected to interact with F-actin, save PFK, PGM, and PK. If any of the expected interactions are not observed, we must again be concerned about the effects of the fusion construction. The fluorescent protein tags may interfere with the proper folding of the enzymes, or may block the interaction face with F-actin. The problem may be resolved by switching the position of the fluorescent protein tag on either protein (N or C-terminus), or by adjusting the linker length.
To ensure that changes in the FRAP recovery rates are the consequence of specific interactions between the glycolytic enzymes and F-actin, CFP and YFP will be used as negative controls. To further verify the specificity, the previously mentioned Hog1-fluoresent protein tagged construct can also be used as a negative control. All glycolytic enzymes are expected to interact with F-actin, save PFK, PGM, and PK. If any of the expected interactions are not observed, we must again be concerned about the effects of the fusion construction. The fluorescent protein tags may interfere with the proper folding of the enzymes, or may block the interaction face with F-actin. The problem may be resolved by switching the position of the fluorescent protein tag on either protein (N or C-terminus), or by adjusting the linker length.
The effect of F-actin on glycolytic enzymes interactions in vitro
To test the hypothesis that F-actin stabilizes the protein-protein interactions of the glycolytic metabolon, the FRET-based in vitro binding experiment will be repeated in the presence of F-actin. As before, the acceptor-tagged protein will be titrated into a solution of the donor-tagged protein. This experiment will allow comparison of apparent dissociation constants with and without F-actin. In accordance with the proposed model (Fig. 2), it is expected that all protein-protein interactions will have higher stability in the presence of F-actin, save those of PFK, PGM, and PK. For these, we expect no difference in affinity. The same negative controls will be done as before in the absence of F-actin.
Glycolytic enzyme protein-protein interactions in vivo
Binary interactions that can be detected in vitro by FRET will be validated in vivo. Using gene targeting by homologous recombination, the genes of the yeast chromosome corresponding to the proteins of interest can be modified to incorporate sequences encoding the fluorescent protein tags. Provided that these fusion proteins properly function in the glycolytic metabolon, the resulting cells are expected to be viable and show a functional glycolytic phenotype (growth given glucose as a carbon source). If so, fluorescence microscopy should allow detection of the FRET interaction, providing confirmation that the proteins associate in vivo.
Next, the role of F-actin in the stability of the glycolytic complex can be probed in vivo using the validated protein-protein interactions. After observing an in vivo interaction by FRET, the cells can be treated with cytochalasin D. This compound inhibits actin polymerization, resulting in the rapid disintegration of F-actin filaments [5]. If F-actin is essential for the metabolon’s stability, we expect dissociation of the complex and abolition of the FRET signal (Fig. 4). After a sufficient washout period to allow clearance of the drug, the FRET signal is expected to return concomitantly with the reformation of F-actin and the metabolon complex. This process can also be monitored by FRAP. Yeast cells modified with one fluorescent protein-tagged glycolytic enzyme will be used. The tagged enzyme is expected to have an attenuated diffusion rate and fluorescence recovery while bound in a metabolon complex. As before, the cells can then be treated with cytochalasin D to disrupt the F-actin polymers. Given that this reduces the stability of the metabolon complex and causes it to dissociate, an increase in the diffusion rate and fluorescence recovery rate of the tagged enzyme is expected.
Next, the role of F-actin in the stability of the glycolytic complex can be probed in vivo using the validated protein-protein interactions. After observing an in vivo interaction by FRET, the cells can be treated with cytochalasin D. This compound inhibits actin polymerization, resulting in the rapid disintegration of F-actin filaments [5]. If F-actin is essential for the metabolon’s stability, we expect dissociation of the complex and abolition of the FRET signal (Fig. 4). After a sufficient washout period to allow clearance of the drug, the FRET signal is expected to return concomitantly with the reformation of F-actin and the metabolon complex. This process can also be monitored by FRAP. Yeast cells modified with one fluorescent protein-tagged glycolytic enzyme will be used. The tagged enzyme is expected to have an attenuated diffusion rate and fluorescence recovery while bound in a metabolon complex. As before, the cells can then be treated with cytochalasin D to disrupt the F-actin polymers. Given that this reduces the stability of the metabolon complex and causes it to dissociate, an increase in the diffusion rate and fluorescence recovery rate of the tagged enzyme is expected.
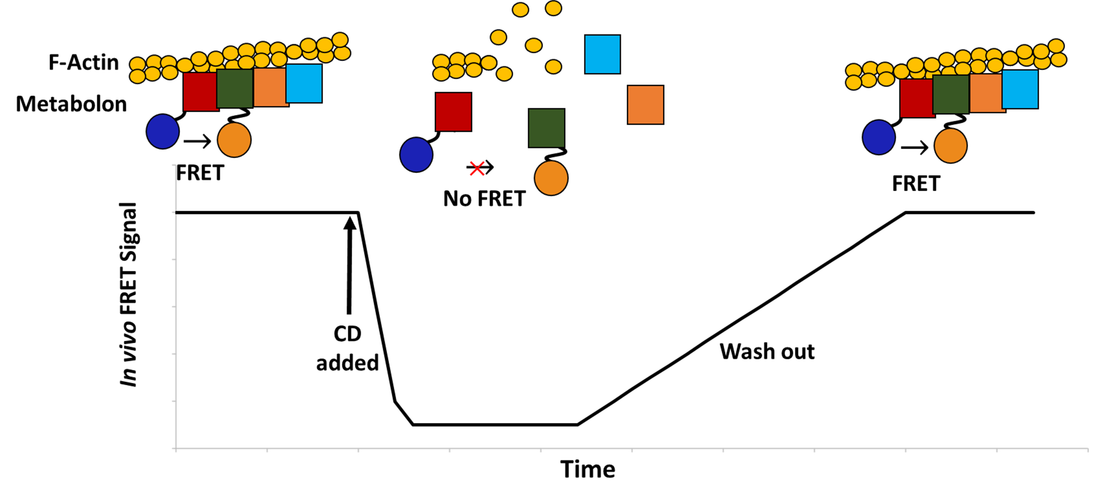
Figure 4. Expected in vivo FRET response over time during the experiment using cytochalasin D (CD).
As with the in vitro experiments, negative controls are essential. Yeast cells will be transformed with a plasmid encoding YFP and CFP and used as a negative control. This will ensure that YFP and CFP do not associate non-specifically in the cellular environment, and that the effect of cytochalasin D in the FRAP experiment is specific. Additionally, the Hog1 yeast chromosome gene will also be tagged with a fluorescent protein and tested for interaction with the glycolytic enzymes as a negative control in the in vivo FRET experiments. A possible limitation of using FRET in vivo is its sensitivity. Detecting interactions in vivo with dissociation constants greater than 10 μM is likely unfeasible [11]. Thus, it will be most efficient to focus in vivo work on protein-protein interactions found to have sufficient affinity in vitro. A few in vivo interactions will be enough to show the existence of the metabolon and elucidate the role of F-actin. It would be possible to detect low affinity interactions in vivo with a more sensitive technique, like bimolecular fluorescence complementation [8]. However, this technique is undesirable due to its irreversibility, which would preclude many of the above experiments.
As with the in vitro experiments, negative controls are essential. Yeast cells will be transformed with a plasmid encoding YFP and CFP and used as a negative control. This will ensure that YFP and CFP do not associate non-specifically in the cellular environment, and that the effect of cytochalasin D in the FRAP experiment is specific. Additionally, the Hog1 yeast chromosome gene will also be tagged with a fluorescent protein and tested for interaction with the glycolytic enzymes as a negative control in the in vivo FRET experiments. A possible limitation of using FRET in vivo is its sensitivity. Detecting interactions in vivo with dissociation constants greater than 10 μM is likely unfeasible [11]. Thus, it will be most efficient to focus in vivo work on protein-protein interactions found to have sufficient affinity in vitro. A few in vivo interactions will be enough to show the existence of the metabolon and elucidate the role of F-actin. It would be possible to detect low affinity interactions in vivo with a more sensitive technique, like bimolecular fluorescence complementation [8]. However, this technique is undesirable due to its irreversibility, which would preclude many of the above experiments.
Conclusion/Significance
Glycolysis is a metabolic pathway fundamental to the chemistry of life. It is essential for both aerobic and anaerobic respiration and is present in most known organisms [3]. The proposed research will inform the in vivo structural organization of this important biological process. Furthermore, insights gained through this project may have implications towards our general understanding of the structure, regulation, and function of metabolons.
An additional consequence of this research will be the development of new tools to study yeast glycolysis in vivo. If successful, the FRET-based approach will provide a versatile in vivo probe for the status of the glycolytic metabolon. It has been proposed that the dynamic association/dissociation of metabolon complexes could be a means to regulate metabolic flux [3]. The technique will permit observation of these dynamic processes in living cells, allowing this hypothesis to be experimentally tested. Given the putative role of F-actin in the glycolytic complex’s stability, it is interesting that yeast F-actin structure and its complement of bound proteins is known to be highly regulated and dynamic [12]. An intriguing hypothesis emerges from this, that F-actin binding could be a biologically relevant means of glycolytic regulation. In the future, this FRET-based technique may allow detailed in vivo experiments correlating glycolytic metabolon association, glycolysis rate, and metabolic state.
An additional consequence of this research will be the development of new tools to study yeast glycolysis in vivo. If successful, the FRET-based approach will provide a versatile in vivo probe for the status of the glycolytic metabolon. It has been proposed that the dynamic association/dissociation of metabolon complexes could be a means to regulate metabolic flux [3]. The technique will permit observation of these dynamic processes in living cells, allowing this hypothesis to be experimentally tested. Given the putative role of F-actin in the glycolytic complex’s stability, it is interesting that yeast F-actin structure and its complement of bound proteins is known to be highly regulated and dynamic [12]. An intriguing hypothesis emerges from this, that F-actin binding could be a biologically relevant means of glycolytic regulation. In the future, this FRET-based technique may allow detailed in vivo experiments correlating glycolytic metabolon association, glycolysis rate, and metabolic state.
References
[1] Srere PA (1987) Complexes of sequential metabolic enzymes. Annu Rev Biochem. 56, 89–124.
[2] Ovadi J and Saks V (2004) On the origin of intracellular compartmentation and organized metabolic systems. Mol Cell Biochem. 256, 5–12.
[3] Menard L, Maughan D and Vigoreaux J (2014) The Structural and Functional Coordination of Glycolytic Enzymes in Muscle: Evidence of a Metabolon? Biology. 3, 623-644.
[4] Puchulu-Campanella E, Chu H, Anstee D, Galan J, Tao A and Low P (2013) Identification of the Components of a Glycolytic Enzyme Metabolon on the Human Red Blood Cell Membrane. J Biol Chem. 288, 848-858.
[5] Araiza-Olivera D, Chiquetee-Felix N, Rosas-Lemus M, Sampedro J, Pena A, Mujica A and Uribe-Carvajal S (2013) A glycolytic metabolon in Saccharomyces cerevisiae is stabilized by F-actin. FEBS. 280, 3887-3905.
[6] Waingeh VF, Gustafson CD, Kozliak EI, Lowe SL, Knull HR and Thomasson KA (2006) Glycolytic enzyme interactions with yeast and skeletal muscle F-actin. Biophys J. 90, 1371–1384.
[7] Song Y, Madahar V and Liao J (2010) Development of FRET Assay into Quantitative and High-throughput Screening Technology Platforms for Protein–Protein Interactions. Annals Biomed Eng. 39, 124-1234.
[8] Weber-Boyvat M, Li S, Skarp K, Olkkonen V, Yan D and Jäntti J (2015) Bimolecular Fluorescence Complementation (BiFC) Technique in Yeast Saccharomyces cerevisiae and Mammalian Cells. Method Mol Bio. 1270, 277-288.
[9] Fields S and Song O (1989) A novel genetic system to detect protein-protein interactions. Nat Lett. 340, 245-246.
[10] Williamson M and Sutcliffe M (2010) Protein-protein interactions. Biochem Soc Trans. 38, 875-878.
[11] Piehler J (2005) New methodologies for measuring protein interactions in vivo and in vitro. Curr Opin Struct Biol. 15, 4-14.
[12] Mirshra M, Huang J and Balasubramanian M (2014) The yeast actin cytoskeleton. FEMS Microbiol Rev. 38, 213-227.
[2] Ovadi J and Saks V (2004) On the origin of intracellular compartmentation and organized metabolic systems. Mol Cell Biochem. 256, 5–12.
[3] Menard L, Maughan D and Vigoreaux J (2014) The Structural and Functional Coordination of Glycolytic Enzymes in Muscle: Evidence of a Metabolon? Biology. 3, 623-644.
[4] Puchulu-Campanella E, Chu H, Anstee D, Galan J, Tao A and Low P (2013) Identification of the Components of a Glycolytic Enzyme Metabolon on the Human Red Blood Cell Membrane. J Biol Chem. 288, 848-858.
[5] Araiza-Olivera D, Chiquetee-Felix N, Rosas-Lemus M, Sampedro J, Pena A, Mujica A and Uribe-Carvajal S (2013) A glycolytic metabolon in Saccharomyces cerevisiae is stabilized by F-actin. FEBS. 280, 3887-3905.
[6] Waingeh VF, Gustafson CD, Kozliak EI, Lowe SL, Knull HR and Thomasson KA (2006) Glycolytic enzyme interactions with yeast and skeletal muscle F-actin. Biophys J. 90, 1371–1384.
[7] Song Y, Madahar V and Liao J (2010) Development of FRET Assay into Quantitative and High-throughput Screening Technology Platforms for Protein–Protein Interactions. Annals Biomed Eng. 39, 124-1234.
[8] Weber-Boyvat M, Li S, Skarp K, Olkkonen V, Yan D and Jäntti J (2015) Bimolecular Fluorescence Complementation (BiFC) Technique in Yeast Saccharomyces cerevisiae and Mammalian Cells. Method Mol Bio. 1270, 277-288.
[9] Fields S and Song O (1989) A novel genetic system to detect protein-protein interactions. Nat Lett. 340, 245-246.
[10] Williamson M and Sutcliffe M (2010) Protein-protein interactions. Biochem Soc Trans. 38, 875-878.
[11] Piehler J (2005) New methodologies for measuring protein interactions in vivo and in vitro. Curr Opin Struct Biol. 15, 4-14.
[12] Mirshra M, Huang J and Balasubramanian M (2014) The yeast actin cytoskeleton. FEMS Microbiol Rev. 38, 213-227.
 RSS Feed
RSS Feed