Ibuprofen (brand name Advil or Motrin) is one of the most widely used nonsteroidal anti-inflammatory drugs (NSAIDs). It is useful for reducing pain, inflammation, and fever. In this article I examine the total synthesis of ibuprofen from benzene. I review two classic industrial organic syntheses of ibuprofen, the Boots process and the Boots-Hoechst-Celanese process (abbreviated BHC or Hoechst). I use benzene as a starting point to demonstrate how complex structural elaboration of a simple precursor is possible in organic synthesis. In addition to discussing these synthetic routes, I clarify all mechanisms with arrow-pushing diagrams. To appraise the synthetic challenges of preparing ibuprofen, we begin with a retrosynthetic analysis.
Retrosynthetic analysis
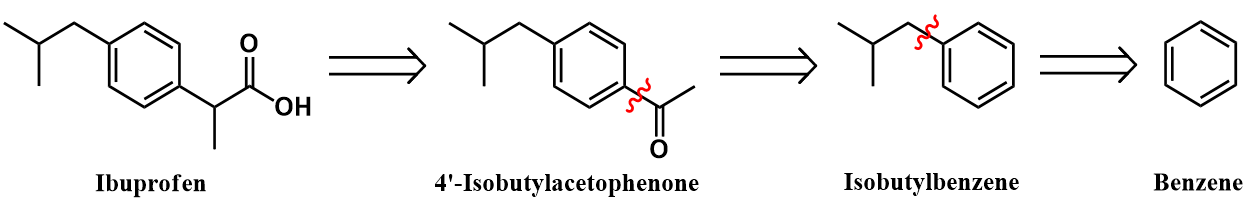
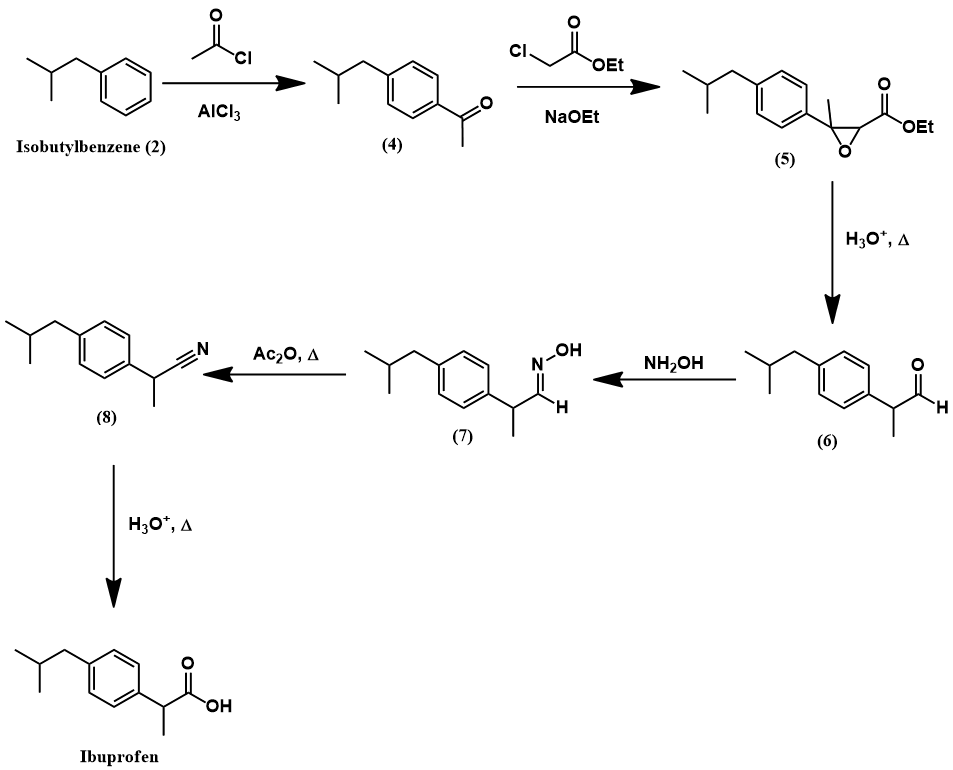
Both the Boots and the Hoechst processes access ibuprofen through the same key intermediate, 4’-isobutylacetophenone. This is the most challenging aspect of the synthesis, where the para-acetyl group of this compound must be transformed to a propionic acid moiety. These processes achieve this transformation through rather different strategies, as we will soon see. However, both methods produce this key intermediate through a common approach: a Friedel-Crafts acylation of isobutylbenzene. Finally, this compound must, in turn, be prepared from benzene. I outline a synthesis of isobutylbenzene below.
Synthesis of isobutylbenzene from benzene
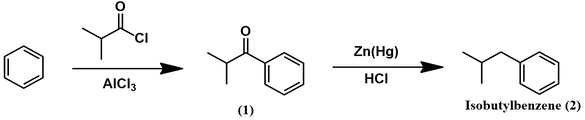
First, a Friedel-Crafts acylation functionalizes benzene with an isobutyryl moiety (1). This substituent is reduced to an isobutyl group through a Clemmensen reduction, yielding isobutylbenzene (2).
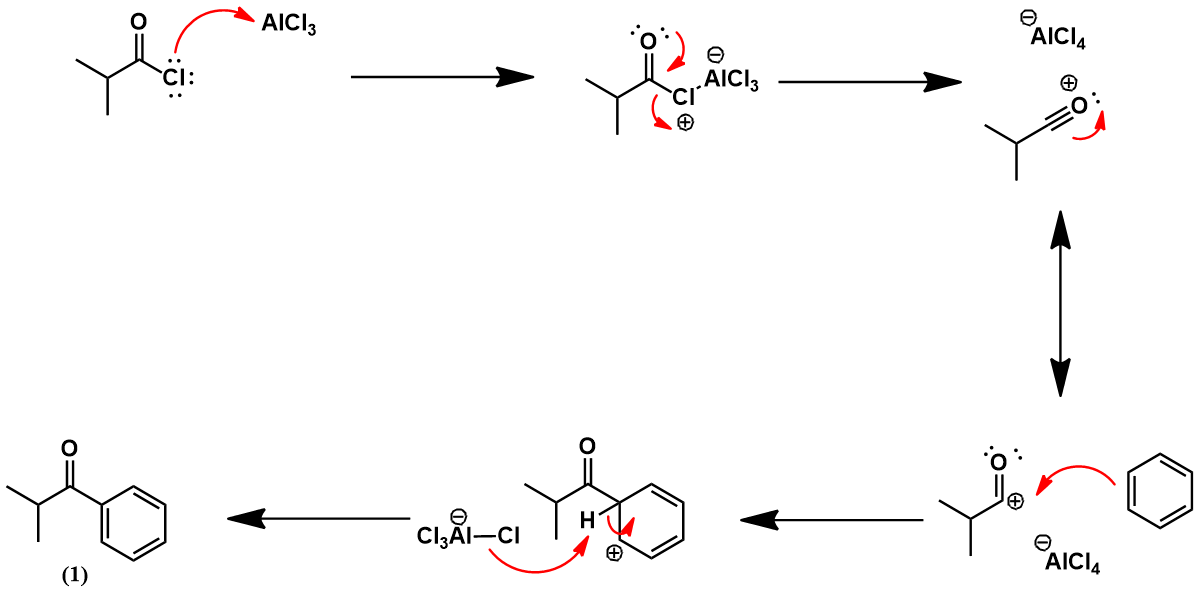
Mechanistically, the Friedel-Crafts acylation is a kind of electrophilic aromatic substitution. It begins with activation of the acid chloride through the Lewis acidity of aluminium trichloride (AlCl3). This stabilizes an acylium ion of the acetylating agent, which is a potent electrophile. Nucleophilic attack by benzene occurs at this center, yielding the acylated product (1). This reaction proceeds through a positively charged arenium ion intermediate (sigma-complex). Conveniently, the product’s substituent is electron-withdrawing, limiting the formation of undesired, multiply substituted products. It is unreactive toward further electrophilic substitution because it is less able to stabilize the positively charged arenium intermediate.
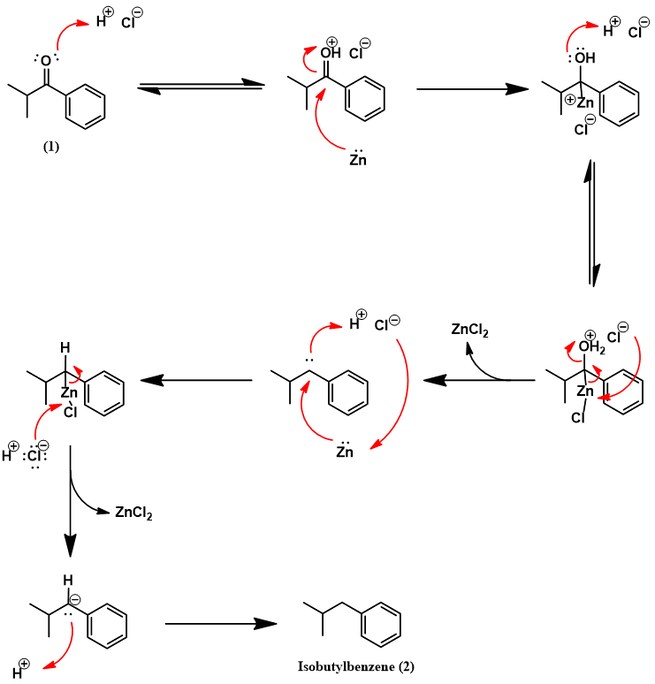
The ketone of (1) is then removed to afford isobutylbenzene by Clemmensen reduction. Though a classic reaction, its mechanism has not yet been fully elucidated. The two main mechanistic proposals proceed through carbanionic or carbene species, with some experimental evidence supporting either. Recently, Sanchez-Viesca and coworkers suggested a new mechanism which coherently invokes both types of species [1]:
This approach for isobutylbenzene synthesis may seem overly complicated, since direct alkylation of benzene is possible through Fiedel-Crafts alkylation. It is tempting to envision directly alkylating with isobutyl chloride to attain the product in a single step. However, this approach doesn’t work. Instead of getting isobutylbenzene, sec-butylbenzene (3) would be the major product.
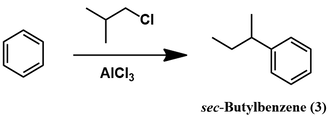
The reason for this is clarified by careful consideration of the reaction’s mechanism. Activation of isobutyl chloride with a strong Lewis acid (AlCl3) generates a primary carbocation. This highly unstable intermediate rapidly undergoes an intramolecular rearrangement to the more stable secondary carbocation. This is the main alkylating species which reacts with benzene, giving the undesired sec-butyl product.
Another disadvantage of the Friedel-Crafts alkylation is that unlike for the Friedel-Crafts acylation, the product is more nucleophilic than the starting material. There is therefore a tendency to form to multiply alkylated side products. This is a consequence of alkyl substituents being weakly electron-donating, affording better stabilization of the arenium ion intermediate.
Synthesis of ibuprofen by the Boots process [2]
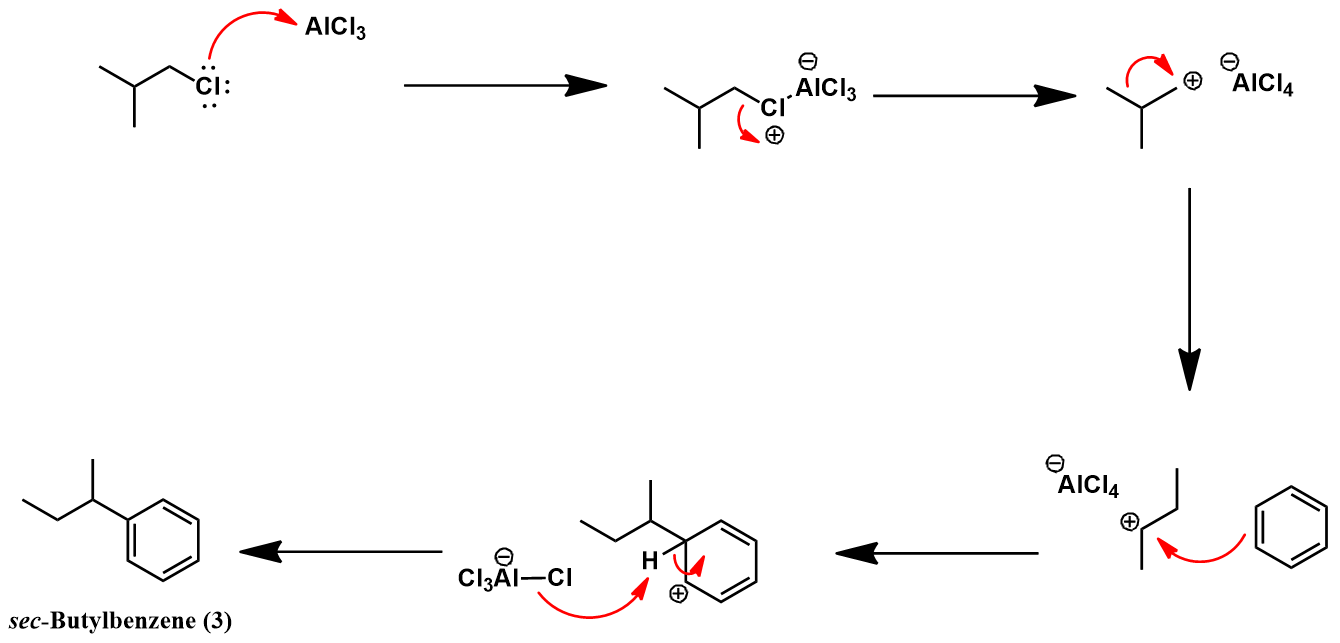
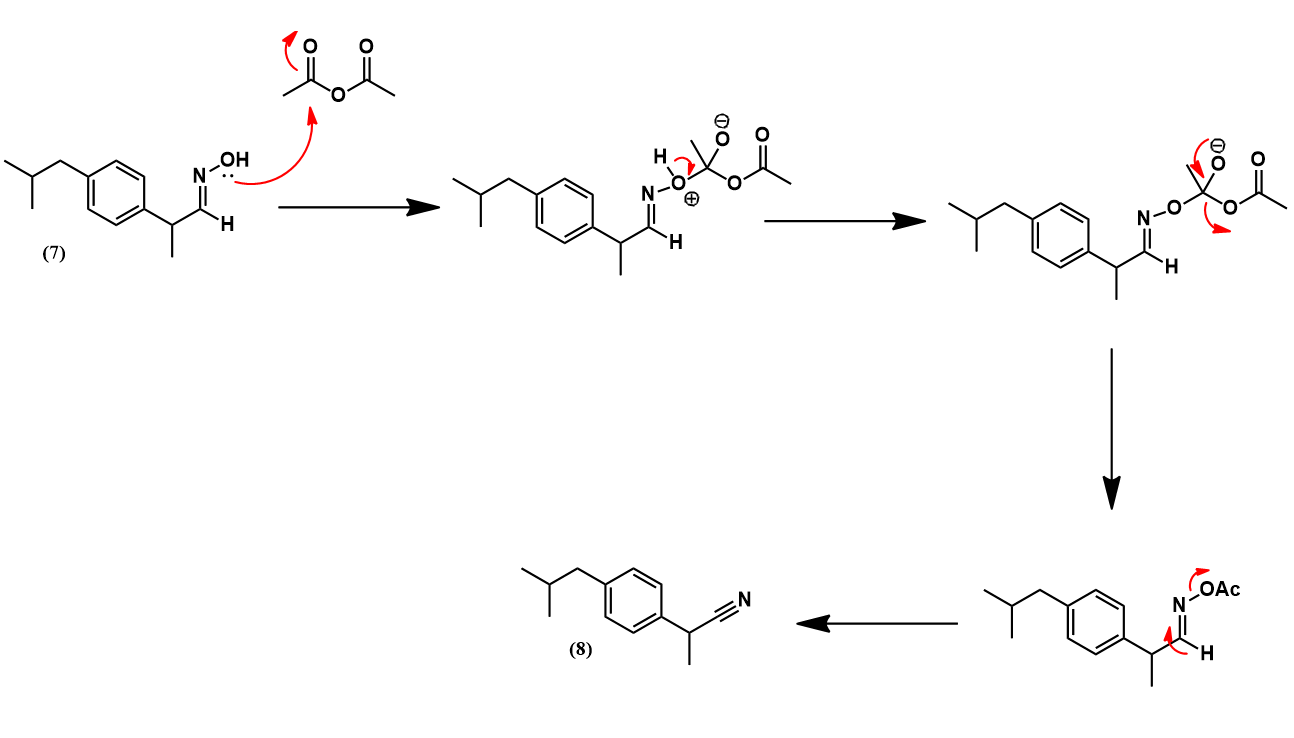
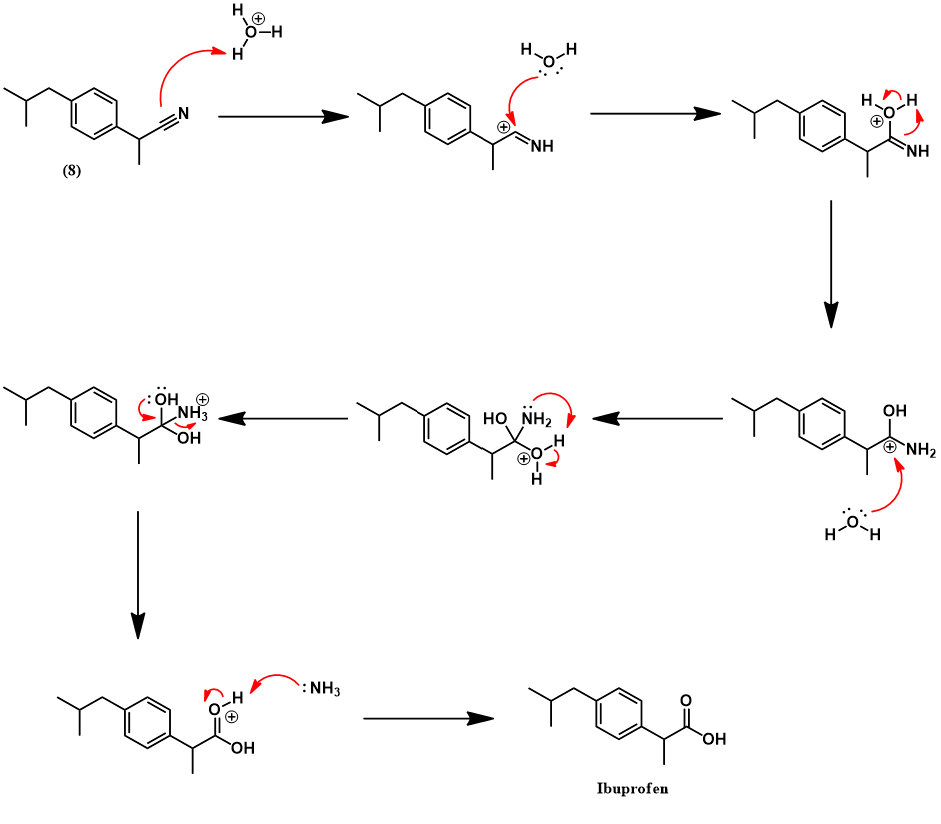
Having prepared isobutylbenzene, we now review the historic synthesis of ibuprofen developed the Boots company. This is the company which originally discovered the analgesic activity of this compound. A full synthetic scheme is presented below. The synthesis begins with another Friedel-Crafts acylation on isobutylbenzene with acetyl chloride to give (4). The next synthetic steps work to convert this compound’s para-acetyl group to ibuprofen’s propionic acid substituent. First, The acetyl group is converted to an α,β-epoxy ester (5) via a Darzens reaction with ethyl chloroacetate. Next, hydrolysis and decarboxylation give the aldehyde (6). Condensation with hydroxylamine affords an aldoxime (7) which is then dehydrated to the nitrile (8) using acetic anhydride. Lastly, acid-catalyzed hydrolysis of the nitrile to a carboxylic acid yields ibuprofen.
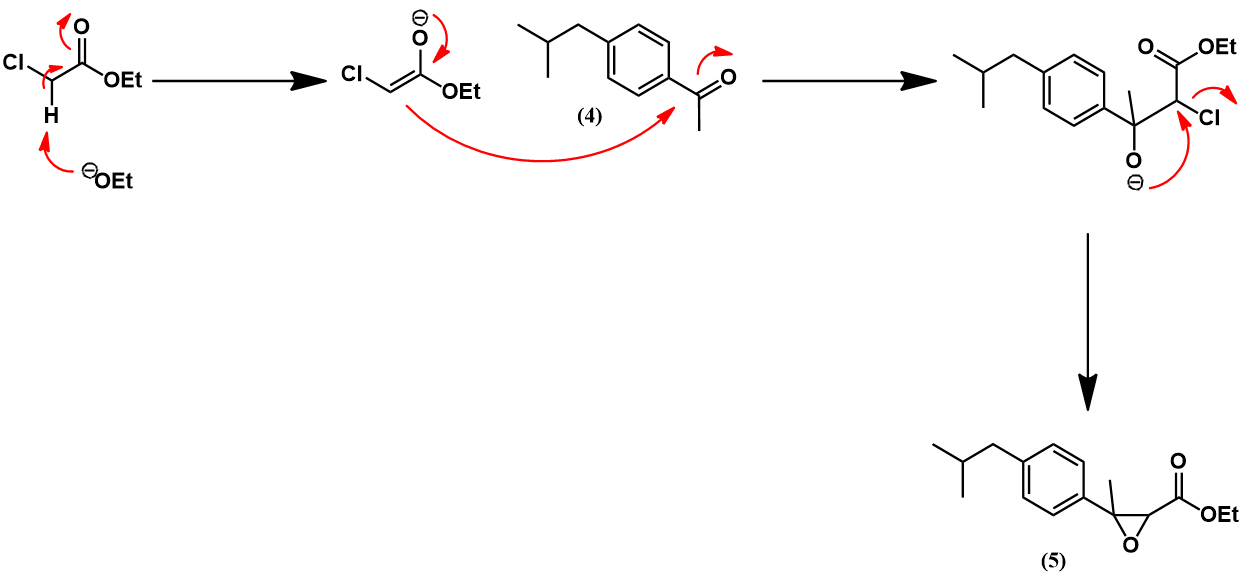
The acylation of isobutylbenzene (2) to 4’-isobutylacetophenone (4) occurs via a Friedel-Crafts acylation, mechanistically identical to the previously shown example. Selectivity for para regiochemistry is achieved because the isobutyl group is othro/para-directing, being weakly electron-donating. Para addition is preferred over ortho due to the steric bulk of the isobutyl group. The next step is a Darzens reaction which transforms the acetyl substituent to an α,β-epoxy ester. The mechanism begins with deprotonation of ethyl chloroacetate at its halogenated position, forming a carbanion stabilized by resonance with its enolate form. This nucleophile attacks at the carbonyl carbon of (4). The oxygen anion of the resulting tetrahedral intermediate then participates in an intramolecular SN2 reaction, forming the α,β-epoxy ester (5) with expulsion of the chloride leaving group.
The next reaction converts the α,β-epoxy ester (5) to an aldehyde. First, acid-catalyzed hydrolysis of the ester occurs, affording the carboxylic acid. Under the acidic conditions this species spontaneously decarboxylates to give the aldehyde (6) in equilibrium with its enol tautomer.
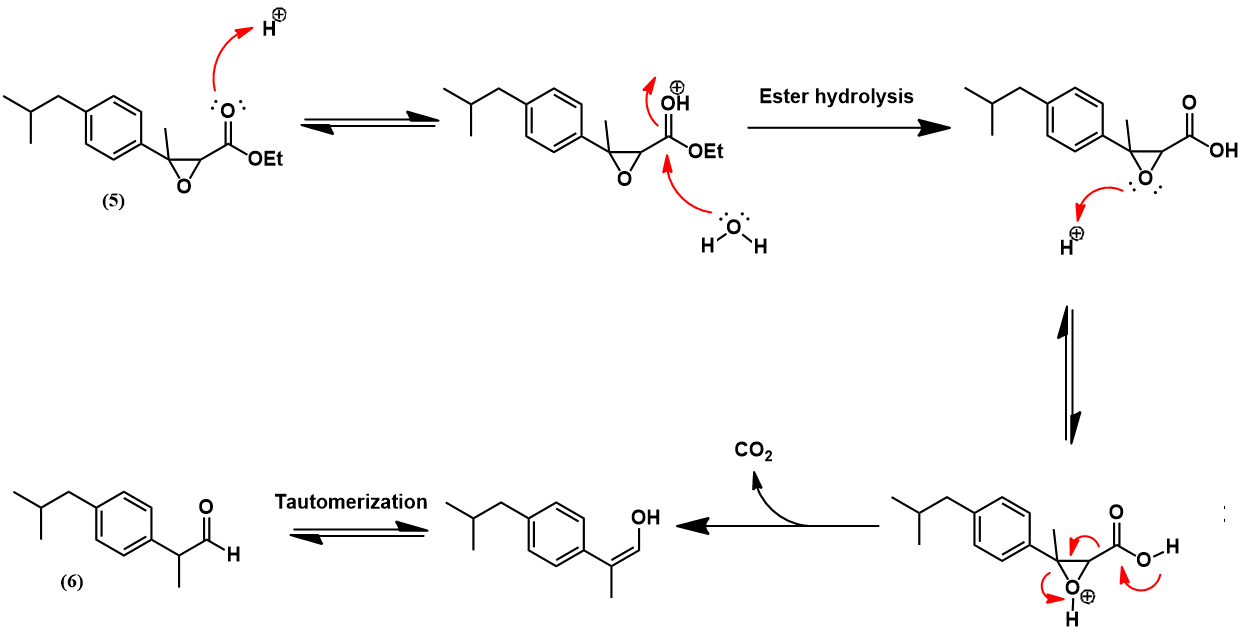
Hydroxylamine is then condensed with aldehyde (6) to give the aldoxime (7). Mechanistically, this begins with nucleophilic attack of hydroxylamine at the aldehyde carbonyl carbon. After a few proton transfers, expulsion of a water molecule leaves the aldoxime.
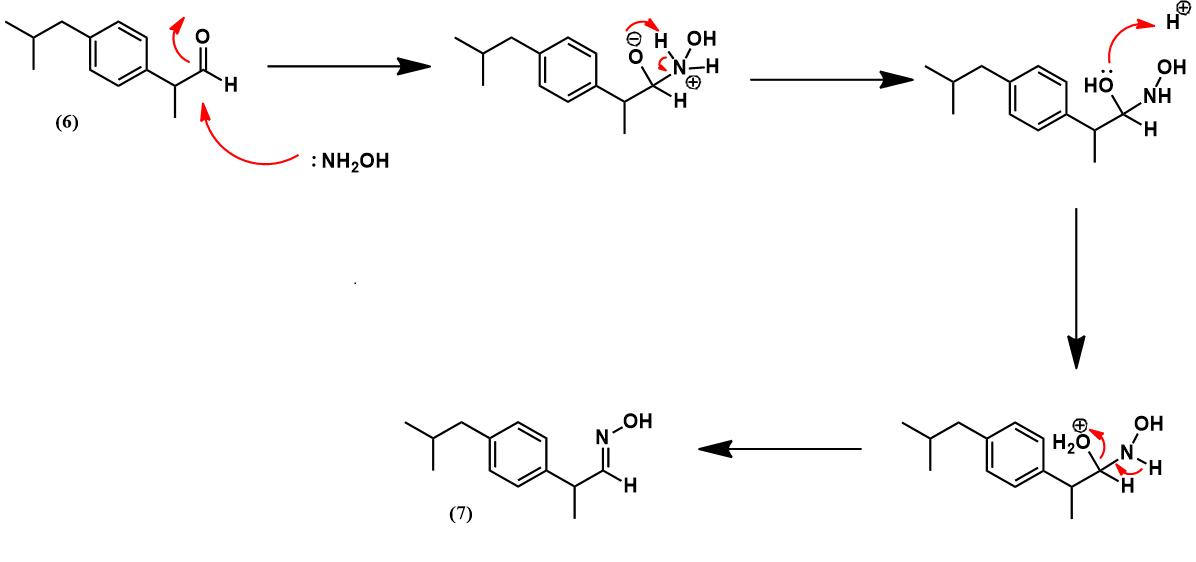
The aldoxime (7) is then dehydrated with acetic anhydride. Acetylation of (7) gives an O-acetyl aldoxime which readily eliminates acetic acid to give the nitrile product (8).
Finally, the nitrile (8) is hydrolyzed under acid-catalyzed conditions. Two nucleophilic additions of water and elimination of ammonium give the carboxylic acid (ibuprofen).
The Boots-Hoechst-Celanese ibuprofen synthesis [3]
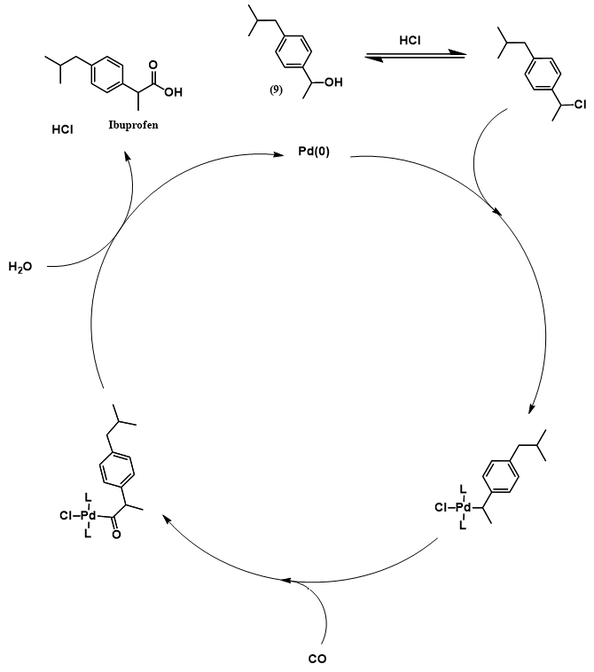
The Hoechst process for ibuprofen synthesis, developed by the Boots-Hoechst-Celanese company, was a significant improvement over the original Boots process. A synthetic scheme for this route follows. Though different reaction conditions are used, it begins like the Boots process with a Friedel-Crafts acylation of isobutylbenzene to make 4’-isobutylacetophenone (4). This route differs dramatically in how this intermediate is transformed to ibuprofen, however. First, the aromatic ketone of (4) is reduced to an alcohol (9) via catalytic hydrogenation. Ibuprofen is produced directly from (9) through carbonylation with carbon monoxide in the presence of a palladium catalyst.
The Hoechst method has several advantages over the Boots process, ultimately being a much “greener” pathway. Most obviously, the synthesis is accomplished in fewer steps than the latter. Isobutylbenzene is converted to ibuprofen in just three steps, while the Boots process requires six. The process also has superior atom economy, achieving twice the atom utilization (~80%) of the original Boots process (~40%). Lastly, the process involved efficient isolation and recovery of catalysts and reaction by-products, limiting the waste produced. Because of these remarkable improvements and the importance of ibuprofen synthesis, this route was awarded the EPA’s Presidential Green Chemistry Award [4].
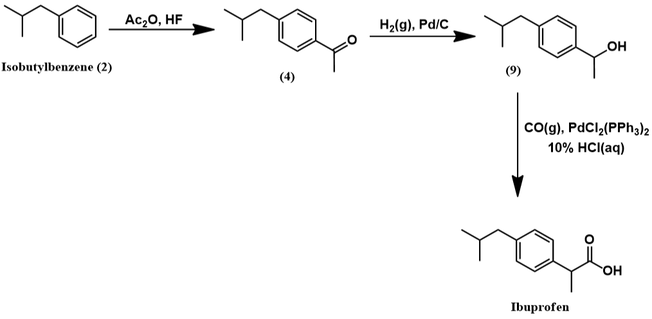
Mechanistically, the Friedel-Crafts acylation of isobutylbenzene to (4) proceeds in identical fashion as shown earlier for the Boots process. However, HF replaces the Lewis acid catalyst and acetic anhydride replaces acetyl chloride. The next synthetic step is a pallidum-catalyzed hydrogenation of aromatic ketone (4) to alcohol (9). The palladium catalyst facilitates the reaction through activation of H2 by surface adsorption. The ketone also adsorbs to the palladium surface [5]. This brings it in proximity to the activated H2 and promotes hydrogenation. These steps are represented schematically below.
Mechanistically, the Friedel-Crafts acylation of isobutylbenzene to (4) proceeds in identical fashion as shown earlier for the Boots process. However, HF replaces the Lewis acid catalyst and acetic anhydride replaces acetyl chloride. The next synthetic step is a pallidum-catalyzed hydrogenation of aromatic ketone (4) to alcohol (9). The palladium catalyst facilitates the reaction through activation of H2 by surface adsorption. The ketone also adsorbs to the palladium surface [5]. This brings it in proximity to the activated H2 and promotes hydrogenation. These steps are represented schematically below.
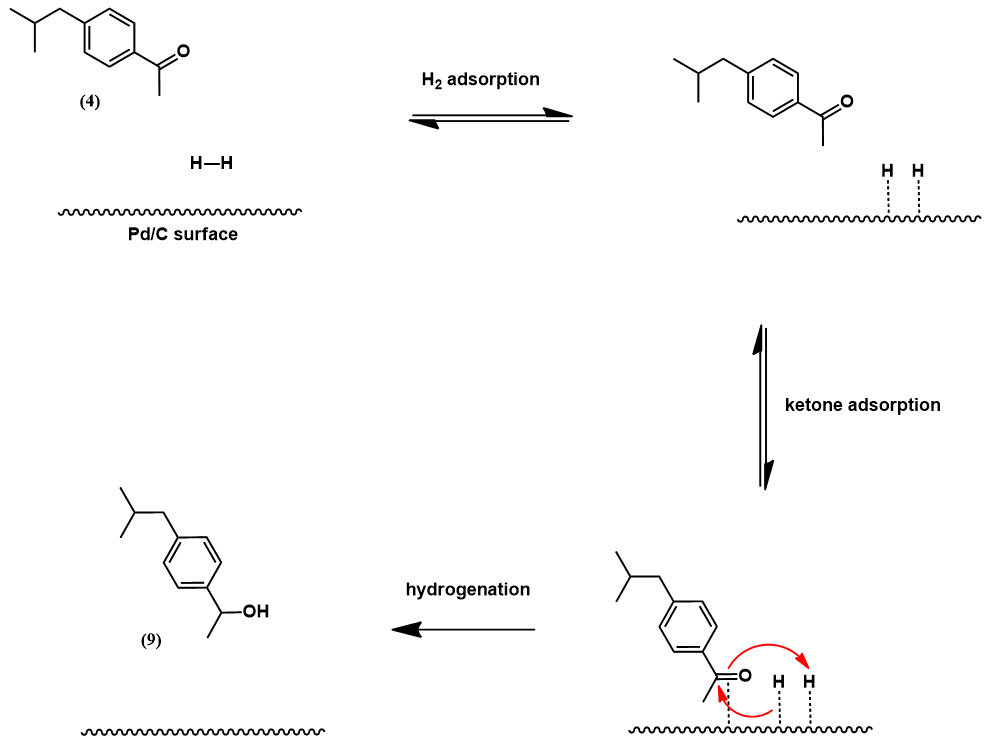
The final step is carbonylation of alcohol (9) with carbon monoxide, another palladium-catalyzed reaction. A proposed mechanism is presented below [6]. In the presence of abundant hydrochloric acid, alcohol (9) is in equilibrium with its benzyl chloride analog. The catalytic cycle begins with oxidative addition of the benzyl chloride to the palladium center. This is followed by insertion of carbon monoxide into the Pd-C sigma bond. Lastly, nucleophilic attack of the acyl intermediate by water releases ibuprofen and regenerates the catalyst.
 RSS Feed
RSS Feed