The range of temperature over which life on Earth thrives is incredible. The majority of organisms are mesophilic, having optimum growth rate at moderate temperature (20 to 45 °C). In contrast, thermophiles have fastest growth from 50 to 70 °C (Reed et al. 2013). Hyperthermophiles have optimum growth as high as 105 °C. At these extreme temperatures, the proteins of mesophilic organisms would certainly be denatured. Thus, to grow and survive at these temperatures thermophiles’ and hyperthermophiles’ proteins must be adapted for stability at high temperature. In this article I review what is known about how these proteins achieve this remarkable stability. Understanding the mechanisms responsible for their thermostability is not just of academic interest: there are a variety of industrial applications (Reed et al., 2013). For instance, understanding these mechanisms may allow the enhancement of commercially important enzymes’ thermostability through protein engineering.
A variety of strategies are used by researchers to elucidate the basis of thermophilic proteins’ thermostability. There are three common approaches. Firstly, large data sets of solved structures or amino acid sequences of thermophilic proteins and homologous mesophilic proteins are compared. Many of the differences between these groups are likely the result of thermophilic adaptions to high temperature. Secondly, the effects of specific amino acid substitutions on thermostability are studied using site-directed mutagenesis. This allows the role of specific interactions to be probed. Lastly, in silico simulations of thermophilic proteins allow the free energy resulting from different types of interactions to be calculated and compared. The basis of high thermostability in thermophilic proteins is complex and not yet fully elucidated. However, it is clear that hydrophobic interactions, ion pair networks, disulfide bonds, and amino acid composition are important factors.
Thermodynamics of Protein Stability at High Temperature
Protein folding as a function of temperature can be understood through stability curves, plots of the free energy of unfolding against temperature (Fig. 1). The most important parameter in quantifying protein stability as a function of temperature is Tm, the melting temperature. At this temperature the probabilities of the protein being folded or unfolded are equal and the Gibbs energy change associated with folding is zero (Fig. 2).
A variety of strategies are used by researchers to elucidate the basis of thermophilic proteins’ thermostability. There are three common approaches. Firstly, large data sets of solved structures or amino acid sequences of thermophilic proteins and homologous mesophilic proteins are compared. Many of the differences between these groups are likely the result of thermophilic adaptions to high temperature. Secondly, the effects of specific amino acid substitutions on thermostability are studied using site-directed mutagenesis. This allows the role of specific interactions to be probed. Lastly, in silico simulations of thermophilic proteins allow the free energy resulting from different types of interactions to be calculated and compared. The basis of high thermostability in thermophilic proteins is complex and not yet fully elucidated. However, it is clear that hydrophobic interactions, ion pair networks, disulfide bonds, and amino acid composition are important factors.
Thermodynamics of Protein Stability at High Temperature
Protein folding as a function of temperature can be understood through stability curves, plots of the free energy of unfolding against temperature (Fig. 1). The most important parameter in quantifying protein stability as a function of temperature is Tm, the melting temperature. At this temperature the probabilities of the protein being folded or unfolded are equal and the Gibbs energy change associated with folding is zero (Fig. 2).
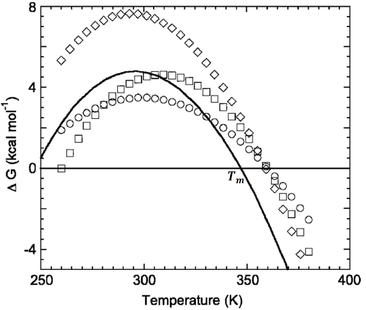
Figure 1. Typical protein stability curves as function of temperature. Comparison of the diamond, circle, and square curves to the solid line visualizes the three thermodynamic mechanisms by which the Tm of a protein can be increased (Razvi and Scholtz, 2006).
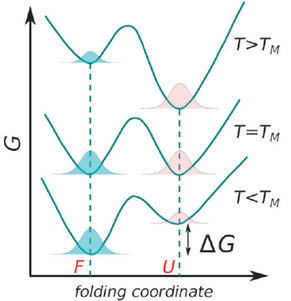
Figure 2. Free energy profile for a two-state model of protein folding (Sterpone and Melchionna, 2011).
The free energy of stabilization of a protein (∆G) as a function of temperature (T) is given by a modified form of the Gibbs-Helmholtz equation (Razvi and Scholtz, 2006):
The free energy of stabilization of a protein (∆G) as a function of temperature (T) is given by a modified form of the Gibbs-Helmholtz equation (Razvi and Scholtz, 2006):
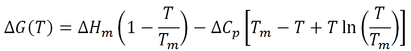
Where Tm is the protein’s melting point, ∆Hm is the change in enthalpy at Tm, and ∆Cp is the change in heat capacity upon unfolding. In order for thermophilic proteins to achieve high thermostability they must have high Tm values. Nojima et al. (1977) suggested three mechanisms by which high Tm values could be attained (Fig. 1). Firstly, raising the entire stability curve by increasing the overall ∆G at all temperatures would increase Tm. The molecular means by which ∆G could be increased includes increasing the number of energetically favorable interactions such as salt bridges, hydrophobic interactions, and hydrogen bonds. Secondly, the stability curve can be broadened by reducing ∆Cp. A decreased ∆Cp corresponds to a more tightly packed core. Lastly, the entire stability curve can be shifted to the right by lowering the entropy change of folding.
Razvi and Scholtz (2006) compared thermodynamic data for 26 sets of thermophilic and mesophilic homolog protein pairs. They found that increased intrinsic stability (higher ∆G at all temperatures) was the most common strategy by which the Tm of thermophilic proteins were increased. Decreasing ∆Cp was the second most common strategy. The least common strategy was decreasing the entropy of folding. Interestingly, thermophilic proteins employ different combinations of these strategies to attain high Tm values. For instance, some used all three strategies, some only the first, the second, the first and third, and so forth. However, in no cases studied was decreasing the entropy of folding used alone as the sole mechanism. Thus, thermodynamically, diverse combinations of strategies are used by thermophilic proteins to attain stability at high temperature.
Razvi and Scholtz (2006) compared thermodynamic data for 26 sets of thermophilic and mesophilic homolog protein pairs. They found that increased intrinsic stability (higher ∆G at all temperatures) was the most common strategy by which the Tm of thermophilic proteins were increased. Decreasing ∆Cp was the second most common strategy. The least common strategy was decreasing the entropy of folding. Interestingly, thermophilic proteins employ different combinations of these strategies to attain high Tm values. For instance, some used all three strategies, some only the first, the second, the first and third, and so forth. However, in no cases studied was decreasing the entropy of folding used alone as the sole mechanism. Thus, thermodynamically, diverse combinations of strategies are used by thermophilic proteins to attain stability at high temperature.
Hydrophobic Interactions
It is reasonable to suspect that hydrophobic interactions play an important role in protein thermostability since the hydrophobic effect increases with temperature (Reed et al., 2013). Indeed, hydrophobic interactions have been demonstrated to be immensely important. This has been observed in a variety of site-directed mutagenesis studies. For instance, Imanaka, Shibazaki, and Takagi (1986) found that amino acid substitutions that increased internal hydrophobicity enhanced the thermostability of proteases. Furthermore, thermophilic proteins generally contain more interactions between hydrophobic residues than mesophilic proteins. Kannan and Vishveshwara (2000) compared thermophilic and mesophilic homolog crystal structures of 24 protein families using a graph spectral method. They found that the majority of thermophilic proteins contained an increased number of aromatic clusters or expanded aromatic networks.
Furthermore, the stabilizing free energy contribution of hydrophobic interactions is greater in thermophilic proteins. Saraboji, Gromiha, and Ponnuswamy (2005) calculated the free energy contributions of a variety of interactions using the structures of thermophilic and mesophilic proteins from 23 different families. In every case they found that the main-chain hydrophobic Gibbs energy was lower in thermophiles than the corresponding mesophilic homologs.
In terms of molecular interactions, increased hydrophobic interactions appear to be one of the most consistent differences between thermophilic and mesophilic proteins. Gromiha et al. (2013) computed and compared parameters relating to hydrophobicity, inter-residue interactions and hydrogen bonds for a dataset of 373 protein families. They found that 80% of thermophilic proteins had greater hydrophobicity than their corresponding mesophilic homologs, making it the strongest predictor of thermophilicity (Fig. 3). Interestingly, the 20% of thermophilic proteins that had decreased hydrophobic interactions all had increased hydrogen bonding or ion pairing. Thus, although the most common, increased hydrophobic interactions is only one of several possible strategies used by thermophilic proteins to achieve stability at high temperature.
It is reasonable to suspect that hydrophobic interactions play an important role in protein thermostability since the hydrophobic effect increases with temperature (Reed et al., 2013). Indeed, hydrophobic interactions have been demonstrated to be immensely important. This has been observed in a variety of site-directed mutagenesis studies. For instance, Imanaka, Shibazaki, and Takagi (1986) found that amino acid substitutions that increased internal hydrophobicity enhanced the thermostability of proteases. Furthermore, thermophilic proteins generally contain more interactions between hydrophobic residues than mesophilic proteins. Kannan and Vishveshwara (2000) compared thermophilic and mesophilic homolog crystal structures of 24 protein families using a graph spectral method. They found that the majority of thermophilic proteins contained an increased number of aromatic clusters or expanded aromatic networks.
Furthermore, the stabilizing free energy contribution of hydrophobic interactions is greater in thermophilic proteins. Saraboji, Gromiha, and Ponnuswamy (2005) calculated the free energy contributions of a variety of interactions using the structures of thermophilic and mesophilic proteins from 23 different families. In every case they found that the main-chain hydrophobic Gibbs energy was lower in thermophiles than the corresponding mesophilic homologs.
In terms of molecular interactions, increased hydrophobic interactions appear to be one of the most consistent differences between thermophilic and mesophilic proteins. Gromiha et al. (2013) computed and compared parameters relating to hydrophobicity, inter-residue interactions and hydrogen bonds for a dataset of 373 protein families. They found that 80% of thermophilic proteins had greater hydrophobicity than their corresponding mesophilic homologs, making it the strongest predictor of thermophilicity (Fig. 3). Interestingly, the 20% of thermophilic proteins that had decreased hydrophobic interactions all had increased hydrogen bonding or ion pairing. Thus, although the most common, increased hydrophobic interactions is only one of several possible strategies used by thermophilic proteins to achieve stability at high temperature.
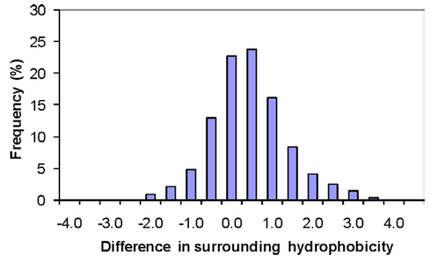
Figure 3. Frequency of thermophilic–mesophilic pairs by difference in surrounding hydrophobicity (Gromiha et al., 2013).
Disulfide Bonds
Disulfide bonds can have significant contributions to the thermostability of thermophilic proteins. Cacciapuoti et al. (2012) demonstrated this for the hyperthermophilic protein 5′-deoxy-5′-methylthioadenosine phosphorylase II. This protein contains a CXC motif (C259/C261) and two intrasubunit disulfide bonds (C138-C205 and C200-C262). The Tm of the wild-type protein was greater than 108 °C, while the C262S and C259S/C261S mutants had Tm values of 102 and 99 °C respectively. Hence, the contribution of the C200-C262 disulfide bond to the protein’s thermostability is large. Furthermore, the CXC motif has been shown to form a strained 11-atom disulfide ring which can act as a reducing agent (Woycechowsky and Raines, 2003). The CXC motif is therefore important in maintaining the native disulfide linkages. This accounts for the even lower Tm of the C259S/C261S double mutant. The importance of disulfide bonds is clear: their contribution is considerable and this protein has even evolved an elaborate mechanism to maintain these linkages.
Disulfide bonds may be particularly important for the stability of quaternary structure. Boutz et al. (2007) performed a proteomic analysis on disulfide linkages in Pyrobaculum aerophilum, a hyperthermophilic archaeon. Disulfide bonds were found to be important for the stability of a variety of protein-protein complexes. They obtained a crystal structure for one of these complexes, a citrate synthase homdimer. Each subunit contained a single intramolecular disulfide bond, cyclizing the protein chain. Remarkably, this cyclization occurred such that the chains of each subunit were topologically interlinked, holding the dimer together (Fig. 4).
Disulfide Bonds
Disulfide bonds can have significant contributions to the thermostability of thermophilic proteins. Cacciapuoti et al. (2012) demonstrated this for the hyperthermophilic protein 5′-deoxy-5′-methylthioadenosine phosphorylase II. This protein contains a CXC motif (C259/C261) and two intrasubunit disulfide bonds (C138-C205 and C200-C262). The Tm of the wild-type protein was greater than 108 °C, while the C262S and C259S/C261S mutants had Tm values of 102 and 99 °C respectively. Hence, the contribution of the C200-C262 disulfide bond to the protein’s thermostability is large. Furthermore, the CXC motif has been shown to form a strained 11-atom disulfide ring which can act as a reducing agent (Woycechowsky and Raines, 2003). The CXC motif is therefore important in maintaining the native disulfide linkages. This accounts for the even lower Tm of the C259S/C261S double mutant. The importance of disulfide bonds is clear: their contribution is considerable and this protein has even evolved an elaborate mechanism to maintain these linkages.
Disulfide bonds may be particularly important for the stability of quaternary structure. Boutz et al. (2007) performed a proteomic analysis on disulfide linkages in Pyrobaculum aerophilum, a hyperthermophilic archaeon. Disulfide bonds were found to be important for the stability of a variety of protein-protein complexes. They obtained a crystal structure for one of these complexes, a citrate synthase homdimer. Each subunit contained a single intramolecular disulfide bond, cyclizing the protein chain. Remarkably, this cyclization occurred such that the chains of each subunit were topologically interlinked, holding the dimer together (Fig. 4).
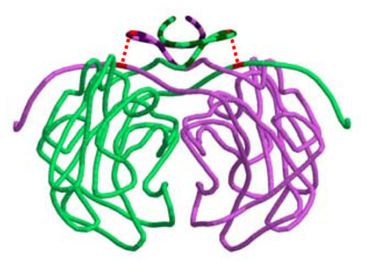
Figure 4. Cartoon representation of Pyrobaculum aerophilum citrate synthase dimer. Dashed red lines depict the two intramolecular disulfide bonds which topologically interlink the subunits (Boutz et al., 2007).
Electrostatic Interactions and Ion Pair Networks
Electrostatic interactions contribute significantly to the thermostability of thermophilic proteins. Although they do often have more salt bridges than mesophilic proteins, the stabilization comes about principally through large ion pair networks (Fig. 5). Yip et al. (1998) studied the relationship between thermal stability and ion pair networks in glutamate dehydrogenases. They used a homology-based modelling approach with known sequences from different species. Lower thermostability correlated with more fragmented ion pair networks.
Electrostatic Interactions and Ion Pair Networks
Electrostatic interactions contribute significantly to the thermostability of thermophilic proteins. Although they do often have more salt bridges than mesophilic proteins, the stabilization comes about principally through large ion pair networks (Fig. 5). Yip et al. (1998) studied the relationship between thermal stability and ion pair networks in glutamate dehydrogenases. They used a homology-based modelling approach with known sequences from different species. Lower thermostability correlated with more fragmented ion pair networks.
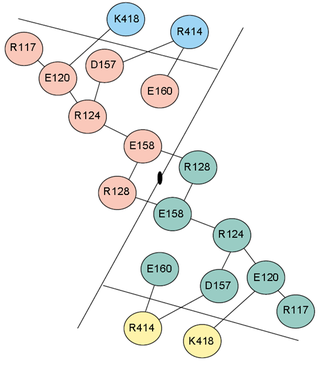
Figure 5. 18-member ion pair network on the dimer interface of glutamate dehydrogenase from Pyrococcus furiosus (Karshikoff and Ladenstein, 2001).
Indeed, there is considerable evidence that the positioning of the ion pairs, rather than the number, is the most energetically important consideration. Xiao and Honig (1999) calculated electrostatic contributions to the folding free energy of hyperthermophilic proteins and their mesophilic homologs. In all pairs studied, they found that the free energy contribution of electrostatic interactions was greater in the hyperthermophilic proteins. Interestingly, the free energy did not correlate well with the number of ionisable residues or ion pairs. Instead, it originated from the optimum placement of charged residues. Bakker, Hunenberger, and McCammon (1999) provided additional evidence that the optimum organization of ion pairs in networks is the principle electrostatic contribution to thermostability. They performed molecular dynamics simulations on the hyperthermophilic protein Sac7d from 300 to 500 K. The majority of electrostatic stabilization came from three large ion pair networks. Despite this, when considered in isolation, the stabilization from individual salt bridges was calculated to be small, some even were even destabilizing. Thus, the energetic stabilization resulting from ion pair networks show a high degree of cooperativity. This accounts for why it is the organization and positioning, rather than the number of ion pairs, which determines the free energy contribution.
Another important finding of Bakker et al. (1999) was that upon raising the simulation temperature from 300 K to 360 K, the protein-solvent interaction energy became less favourable, while the intraprotein interaction energy became more favourable. Both of these effects were principally a consequence of electrostatic contributions. This may be due to decreased desolvation energy, the energetic cost associated with the loss of solvent interactions when a residue becomes buried in a protein. The desolvation energy (∆W) of an ion is given by (Karshikoff and Ladenstein, 2001):
Indeed, there is considerable evidence that the positioning of the ion pairs, rather than the number, is the most energetically important consideration. Xiao and Honig (1999) calculated electrostatic contributions to the folding free energy of hyperthermophilic proteins and their mesophilic homologs. In all pairs studied, they found that the free energy contribution of electrostatic interactions was greater in the hyperthermophilic proteins. Interestingly, the free energy did not correlate well with the number of ionisable residues or ion pairs. Instead, it originated from the optimum placement of charged residues. Bakker, Hunenberger, and McCammon (1999) provided additional evidence that the optimum organization of ion pairs in networks is the principle electrostatic contribution to thermostability. They performed molecular dynamics simulations on the hyperthermophilic protein Sac7d from 300 to 500 K. The majority of electrostatic stabilization came from three large ion pair networks. Despite this, when considered in isolation, the stabilization from individual salt bridges was calculated to be small, some even were even destabilizing. Thus, the energetic stabilization resulting from ion pair networks show a high degree of cooperativity. This accounts for why it is the organization and positioning, rather than the number of ion pairs, which determines the free energy contribution.
Another important finding of Bakker et al. (1999) was that upon raising the simulation temperature from 300 K to 360 K, the protein-solvent interaction energy became less favourable, while the intraprotein interaction energy became more favourable. Both of these effects were principally a consequence of electrostatic contributions. This may be due to decreased desolvation energy, the energetic cost associated with the loss of solvent interactions when a residue becomes buried in a protein. The desolvation energy (∆W) of an ion is given by (Karshikoff and Ladenstein, 2001):
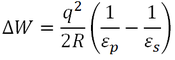
Where q is the ion’s charge, R is its radius, εp is the dielectric constant in the protein, and εs is the dielectric constant of the sovlent. Increasing the temperature decreases the dielectric of water, but has little effect on the dielectric of the protein (Karshikoff and Ladenstein, 2001). Hence, at higher temperatures there is less desolvation energy. This increases the contribution of salt bridges to the free energy of folding at high temperature.
The thermodynamic mechanism by which ion pairs stabilize thermophilic proteins at high temperature is, at least in part, by lowering ∆Cp. Chan, Yu, and Wong (2011) demonstrated this by individually mutating ion pairs with site-directed mutagenesis. Measurement of the ∆Cp of the mutants and comparison to the wild-type protein allowed the contribution of each ion pair to ∆Cp to be calculated. Two ion pairs were identified whose interaction reduced ∆Cp by 0.8 to 1.0 kJ mol-1 K-1.
Amino Acid Composition
A number of changes in the amino acid composition have been noted between thermophilic proteins and their mesophilic homologs. For instance, Kumar, Tasi, and Nussinov (2000) compared 18 non-redundant families of thermophilic and mesophilic proteins. They found that Arg and Tyr frequencies were increased by a significant extent in thermophilic proteins. They argued that these results could be consequences of the increased salt bridges and hydrophobicity commonly associated with thermophilic proteins.
Furthermore, there are statistically significant differences between the amino acid composition of helices in thermophilic and mesophilic proteins. The frequency of Cys and His are decreased, while the frequency of Arg is increased (Kumar, Tasi, and Nussinov, 2000). Interestingly, Arg is a helix favouring residue while Cys, and His are helix disfavouring. In addition, thermophilic proteins have considerably higher helical content in the majority of protein families (Kumar, Tasi, and Nussinov, 2000). It follows that increased helical content and propensity may be an important mechanism of increasing stability at high temperature.
Lastly, Kumar et al. (2000) also found a statistically significant decrease in the proportion of Cys and Ser in thermophilic proteins. This is not surprising: the polar amino acids Asn, Cys, Gln, and Met are considered thermolabile because at high temperature they may undergo deamidation or oxidation reactions (Russel et al. 1997). Thus, decreased usage of these amino acids could be adaption to high temperature, reducing the risk of these undesirable reactions. Although not statistically significant, the proportion of Asn also decreased by a sizeable amount. However, in apparent opposition to this hypothesis the amount of Gln and Met were similar in mesophilic and thermophilic proteins. This is likely because solvent exposure and the local environment are important determinants of an amino acid’s susceptibility to deamidation and oxidation reactions. The biased amino acid usage therefore occurs almost exclusively among the surface residues. A visually striking example of this difference in surface composition is lumazine synthase (Fig. 6). Thus, differences in amino acid composition between thermophilic and mesophilic proteins can be more clearly elucidating by considering the location of each residue. Fukuchi and Nishikawa (2001) compared the interior and exterior amino acid composition of a variety of mesophilic and thermophilic proteins available on the Protein Data Bank. As expected, the frequency of polar uncharged residues (Asn, Ser, and Thr) on the surface of thermophilic proteins was much lower than for mesophilic proteins. Conversely, the proportion of polar charged residues (Asp, Glu, Arg, and Lys) was higher. Although the surface composition is different, Fukuchi and Nishikawa did not find any significant differences in amino acid composition internally.
The thermodynamic mechanism by which ion pairs stabilize thermophilic proteins at high temperature is, at least in part, by lowering ∆Cp. Chan, Yu, and Wong (2011) demonstrated this by individually mutating ion pairs with site-directed mutagenesis. Measurement of the ∆Cp of the mutants and comparison to the wild-type protein allowed the contribution of each ion pair to ∆Cp to be calculated. Two ion pairs were identified whose interaction reduced ∆Cp by 0.8 to 1.0 kJ mol-1 K-1.
Amino Acid Composition
A number of changes in the amino acid composition have been noted between thermophilic proteins and their mesophilic homologs. For instance, Kumar, Tasi, and Nussinov (2000) compared 18 non-redundant families of thermophilic and mesophilic proteins. They found that Arg and Tyr frequencies were increased by a significant extent in thermophilic proteins. They argued that these results could be consequences of the increased salt bridges and hydrophobicity commonly associated with thermophilic proteins.
Furthermore, there are statistically significant differences between the amino acid composition of helices in thermophilic and mesophilic proteins. The frequency of Cys and His are decreased, while the frequency of Arg is increased (Kumar, Tasi, and Nussinov, 2000). Interestingly, Arg is a helix favouring residue while Cys, and His are helix disfavouring. In addition, thermophilic proteins have considerably higher helical content in the majority of protein families (Kumar, Tasi, and Nussinov, 2000). It follows that increased helical content and propensity may be an important mechanism of increasing stability at high temperature.
Lastly, Kumar et al. (2000) also found a statistically significant decrease in the proportion of Cys and Ser in thermophilic proteins. This is not surprising: the polar amino acids Asn, Cys, Gln, and Met are considered thermolabile because at high temperature they may undergo deamidation or oxidation reactions (Russel et al. 1997). Thus, decreased usage of these amino acids could be adaption to high temperature, reducing the risk of these undesirable reactions. Although not statistically significant, the proportion of Asn also decreased by a sizeable amount. However, in apparent opposition to this hypothesis the amount of Gln and Met were similar in mesophilic and thermophilic proteins. This is likely because solvent exposure and the local environment are important determinants of an amino acid’s susceptibility to deamidation and oxidation reactions. The biased amino acid usage therefore occurs almost exclusively among the surface residues. A visually striking example of this difference in surface composition is lumazine synthase (Fig. 6). Thus, differences in amino acid composition between thermophilic and mesophilic proteins can be more clearly elucidating by considering the location of each residue. Fukuchi and Nishikawa (2001) compared the interior and exterior amino acid composition of a variety of mesophilic and thermophilic proteins available on the Protein Data Bank. As expected, the frequency of polar uncharged residues (Asn, Ser, and Thr) on the surface of thermophilic proteins was much lower than for mesophilic proteins. Conversely, the proportion of polar charged residues (Asp, Glu, Arg, and Lys) was higher. Although the surface composition is different, Fukuchi and Nishikawa did not find any significant differences in amino acid composition internally.

Figure 6. Comparison of surface residues in lumazine synthase from Bacillus subtilis (a) and the hyperthermophile Aquifex aeolicus (b). Red and blue represent negatively and positively charged residues respectively. Green and white represent polar uncharged and non-polar residues (Karshikoff and Ladenstein, 2001).
Conclusion
A diverse set of strategies are used by thermophilic proteins to achieve their remarkable thermostability. Thermodynamically, the most common mechanism is to increase the overall stabilizing free energy, ∆G. However, alternative mechanisms, including decreasing ∆Cp and/or entropy of folding are also used by many thermophilic proteins.
A variety of molecular interactions are associated with the high thermostability of thermophilic proteins. The contribution of individual disulfide bonds can be significant, greatly increasing the Tm value. They have been found to be especially important in maintaining quaternary structure at high temperature. In addition, increased hydrophobic interactions are a ubiquitous feature of thermophilic proteins. They have more aromatic clusters and a higher stabilizing free energy contribution from hydrophobic interactions. Large, optimally organized ion pair networks also enhance the thermostability of thermophilic proteins.
Lastly, differences in amino acid composition, particularly on the protein’s surface, contribute to their thermostability. Replacement of uncharged polar surface residues with charged residues protects the protein’s amino acids from potentially detrimental amidation or oxidation reactions. Overall, thermostability is a complex property with contributions from a variety of interactions. Thermophilic proteins use different strategies to variable extents to achieve stability at high temperature.
Conclusion
A diverse set of strategies are used by thermophilic proteins to achieve their remarkable thermostability. Thermodynamically, the most common mechanism is to increase the overall stabilizing free energy, ∆G. However, alternative mechanisms, including decreasing ∆Cp and/or entropy of folding are also used by many thermophilic proteins.
A variety of molecular interactions are associated with the high thermostability of thermophilic proteins. The contribution of individual disulfide bonds can be significant, greatly increasing the Tm value. They have been found to be especially important in maintaining quaternary structure at high temperature. In addition, increased hydrophobic interactions are a ubiquitous feature of thermophilic proteins. They have more aromatic clusters and a higher stabilizing free energy contribution from hydrophobic interactions. Large, optimally organized ion pair networks also enhance the thermostability of thermophilic proteins.
Lastly, differences in amino acid composition, particularly on the protein’s surface, contribute to their thermostability. Replacement of uncharged polar surface residues with charged residues protects the protein’s amino acids from potentially detrimental amidation or oxidation reactions. Overall, thermostability is a complex property with contributions from a variety of interactions. Thermophilic proteins use different strategies to variable extents to achieve stability at high temperature.
References
Bakker PIW, Hunenberger PH and McCammon JA. Molecular dynamics simulations of the hyperthermophilic protein Sac7d from Sulfolobus acidocaldarius: contributions of salt bridges to thermostability. J. Mol. Biol., 1999, 28, 1811–1830.
Boutz DR, Cascio D, Whitelegge J, Perry LJ and Yeates TO. Discovery of a thermophilic protein complex stabilized by topologically interlinked chains. J.Mol. Bio., 2007, 368, 1332–1344.
Cacciapuoti G, Fuccio F, Petraccone L, Del Vecchio P and Percelli M. Role of disulfide bonds in conformational stability and folding of 5’-deoxy-5’-methylthioadenosine phosphorylase II from the hyperthermophilic archaeon Sulfolobus solfataricus. Biochim. Biophy. Acta, 2012, 1824, 1136–1143.
Chan CH, Yu TH and Wong KB. Stabilizing salt-bridge enhances protein thermostability by reducing the heat capacity change of unfolding. PLoS ONE, 2011, 6, 1624.
Fukuchi S and Nishikawa K. Protein surface amino acid compositions distinctively differ between thermophilic and mesophilic bacteria. J. of Mol. Bio., 2001, 309, 835–843.
Gromiha MM, Pathak MC, Saraboji K, Ortlund EA and Gaucher EA. Hydrophobic environment is a key factor for the stability of thermophilic proteins. Proteins, 2013, 81, 715-721.
Imanaka T, Shibazaki M, and Takagi M. A new way of enhancing the thermostability of proteases. Nature, 1986, 324, 695–697.
Kannan N, Vishveshwara S. Aromatic clusters: a determinant of thermal stability of thermophilic proteins. Protein. Eng., 2000, 13, 753–761.
Karshikoff A and Ladenstein R. Ion pairs and the thermotolerance of proteins from hyperthermophiles: a traffic rule for hot roads. Trends Biochem. Sci., 2001, 26, 550–556.
Kumar S, Tsai CJ and Nussinov R. Factors enhancing protein thermostability. Protein Eng., 2000, 13, 179-191.
Nojima H, Ikai A, Oshima T and Noda H. Reversible thermal unfolding of thermostable phosphoglycerate kinase. Thermostability associated with mean zero enthalpy change. J. Mol. Biol., 1977, 116, 429–442.
Razvi A and Scholtz JM. Lessons in stability from thermophilic proteins, Prot. Sci., 2006, 15 1569-1578.
Reed RJ, Lewis HL, Trejo E, Winston V and Evilia C. Protein adaptions in archaeal extremophiles, Archaea, 2013, 2013, 1-14.
Russel RJM, Ferguson JMC, Haugh DW, Danson MJ and Taylor GL. Biochemistry, 1997, 36, 9983-9994.
Saraboji K, Gromiha MM and Ponnuswamy MN. Importance of main chain hydrophobic free energy to the stability of thermophilic proteins. Int. J. Biol. Macromol., 2005, 35, 211–220.
Sterpone F and Melchionna S. Thermophilic proteins: insight and perspective from in silico experiments. Chem. Soc. Rev., 2012, 41, 1665–1676.
Woycechowsky KJ and Raines RT. The CXC motif: a functional mimic of protein disulfide isomerase, Biochemistry, 2013, 42, 5387–5394.
Woycechowsky KJ and Raines RT. The CXC motif: a functional mimic of protein disulfide isomerase. Biochemistry, 2003, 42, 5387–5394.
Xiao L and Honig B. Electrostatic contributions to the stability of hyperthermophilic proteins. J. Mol. Biol., 1999, 289, 1435-1444.
Yip, KSP, Britton KL, Stillman TJ, Lebbink J, Vos WM, Robb FT, et al. Insights into the molecular basis of thermal stability from the analysis of ion pair networks in the glutamate dehydrogenase family. Eur. J. Biochem., 1998, 255, 336–346.
Bakker PIW, Hunenberger PH and McCammon JA. Molecular dynamics simulations of the hyperthermophilic protein Sac7d from Sulfolobus acidocaldarius: contributions of salt bridges to thermostability. J. Mol. Biol., 1999, 28, 1811–1830.
Boutz DR, Cascio D, Whitelegge J, Perry LJ and Yeates TO. Discovery of a thermophilic protein complex stabilized by topologically interlinked chains. J.Mol. Bio., 2007, 368, 1332–1344.
Cacciapuoti G, Fuccio F, Petraccone L, Del Vecchio P and Percelli M. Role of disulfide bonds in conformational stability and folding of 5’-deoxy-5’-methylthioadenosine phosphorylase II from the hyperthermophilic archaeon Sulfolobus solfataricus. Biochim. Biophy. Acta, 2012, 1824, 1136–1143.
Chan CH, Yu TH and Wong KB. Stabilizing salt-bridge enhances protein thermostability by reducing the heat capacity change of unfolding. PLoS ONE, 2011, 6, 1624.
Fukuchi S and Nishikawa K. Protein surface amino acid compositions distinctively differ between thermophilic and mesophilic bacteria. J. of Mol. Bio., 2001, 309, 835–843.
Gromiha MM, Pathak MC, Saraboji K, Ortlund EA and Gaucher EA. Hydrophobic environment is a key factor for the stability of thermophilic proteins. Proteins, 2013, 81, 715-721.
Imanaka T, Shibazaki M, and Takagi M. A new way of enhancing the thermostability of proteases. Nature, 1986, 324, 695–697.
Kannan N, Vishveshwara S. Aromatic clusters: a determinant of thermal stability of thermophilic proteins. Protein. Eng., 2000, 13, 753–761.
Karshikoff A and Ladenstein R. Ion pairs and the thermotolerance of proteins from hyperthermophiles: a traffic rule for hot roads. Trends Biochem. Sci., 2001, 26, 550–556.
Kumar S, Tsai CJ and Nussinov R. Factors enhancing protein thermostability. Protein Eng., 2000, 13, 179-191.
Nojima H, Ikai A, Oshima T and Noda H. Reversible thermal unfolding of thermostable phosphoglycerate kinase. Thermostability associated with mean zero enthalpy change. J. Mol. Biol., 1977, 116, 429–442.
Razvi A and Scholtz JM. Lessons in stability from thermophilic proteins, Prot. Sci., 2006, 15 1569-1578.
Reed RJ, Lewis HL, Trejo E, Winston V and Evilia C. Protein adaptions in archaeal extremophiles, Archaea, 2013, 2013, 1-14.
Russel RJM, Ferguson JMC, Haugh DW, Danson MJ and Taylor GL. Biochemistry, 1997, 36, 9983-9994.
Saraboji K, Gromiha MM and Ponnuswamy MN. Importance of main chain hydrophobic free energy to the stability of thermophilic proteins. Int. J. Biol. Macromol., 2005, 35, 211–220.
Sterpone F and Melchionna S. Thermophilic proteins: insight and perspective from in silico experiments. Chem. Soc. Rev., 2012, 41, 1665–1676.
Woycechowsky KJ and Raines RT. The CXC motif: a functional mimic of protein disulfide isomerase, Biochemistry, 2013, 42, 5387–5394.
Woycechowsky KJ and Raines RT. The CXC motif: a functional mimic of protein disulfide isomerase. Biochemistry, 2003, 42, 5387–5394.
Xiao L and Honig B. Electrostatic contributions to the stability of hyperthermophilic proteins. J. Mol. Biol., 1999, 289, 1435-1444.
Yip, KSP, Britton KL, Stillman TJ, Lebbink J, Vos WM, Robb FT, et al. Insights into the molecular basis of thermal stability from the analysis of ion pair networks in the glutamate dehydrogenase family. Eur. J. Biochem., 1998, 255, 336–346.
 RSS Feed
RSS Feed